ERN-ITHACA clinical practice guidelines and experts consensus statements
Clinical practice guidelines (CPGs) and experts consensus statements (ECSs) are recommendations on how to diagnose and treat a medical condition. They are written by medical experts with the support of patient’s representatives, and are directed to doctors, nurses or any kind of health care professionals.
A CPG summarizes the current medical knowledge on a given disease, weights the benefits and harms of diagnostic procedures and treatments, and give specific recommendations based on this information.
The ITHACA Guideline workgroup works on identifying and subsequently targeting a number of areas where the development and implementation of standardized consensus statements would most benefit the community of patients and families affected by developmental disorders and intellectual diability.
So far, the workgroup has put together 10 groups of multidisciplinary experts to work on the writing of 10 new clinical practice guidelines. 5 of them have already been achieved. They are consultable below:
Clinical practice guidelines and experts consensus statements co-produced by ITHACA
Websites with Guidelines & Consensus Statements available on the Web
In addition to developing these consensus statements, we are also looking at those already in existence in all Member States, how we can endorse their content and translate them, and how we can ensure they reach the widest possible audience. With this in mind, we would like to highlight below some guides which we think you may find useful and that our network will be considering for endorsement in the future.
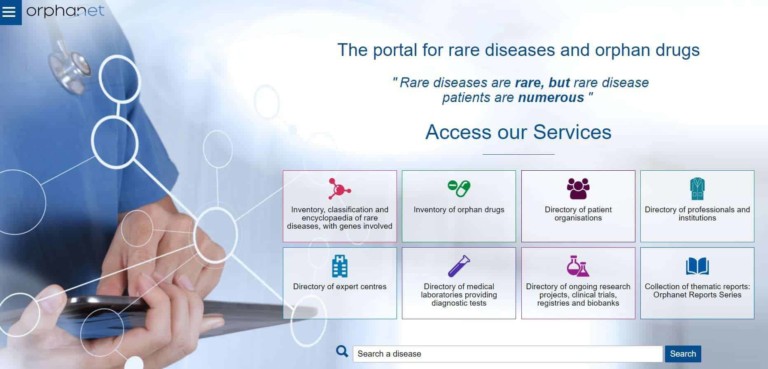
Orphanet
Orphanet is a European website that provides an inventory and classification of rare diseases (RDs), with each entry having its own ORPHA code, an encyclopaedia of expert-authored and peer-reviewed review articles, various clinical guidelines, diagnostic criteria, guidance for genetic testing, disability factsheets etc., and a directory of services (clinics, laboratories, research projects, registries, clinical trials, patient organisations).
ITHACA is a partner of ORPHANET to support RD encyclopaedia and to enrich its entries regarding disorders covered by ITHACA : 72 summaries have been reviewed and updated so far.
Genereviews
GeneReviews is an international point-of-care resource consisting in an online database containing standardized peer-reviewed articles that describe specific heritable diseases. It was established in 1997 as GeneClinics by Roberta A Pagon (University of Washington) with funding from the National Institutes of Health. Its focus is primarily on single-gene disorders, providing current disorder-specific information on diagnosis, management, and genetic counseling. Links to disease-specific and/or general consumer resources are included in each article when available. The database is published on the National Center for Biotechnology Information (NCBI) Bookshelf site. Each chapter is written by one or more experts on the specific condition or disease and goes through a rigorous editing and peer review process before being published online. Articles are updated every two or three years or as needed, and revised whenever significant changes in clinically relevant information occur. Articles are searchable by author, title, gene, and name of disease or protein, and are available free of charge.
Genereviews provides clinically relevant and medically actionable information for inherited conditions in a standardized journal-style format, covering diagnosis, management, and genetic counselling for patients and their families. GeneReviews currently comprises almost 800 chapters, that follow two general formats: 1) chapters focused on a single gene or phenotype ; 2) overviews summarizing genetic causes of common conditions.